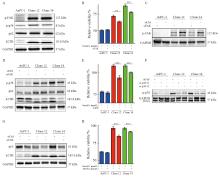
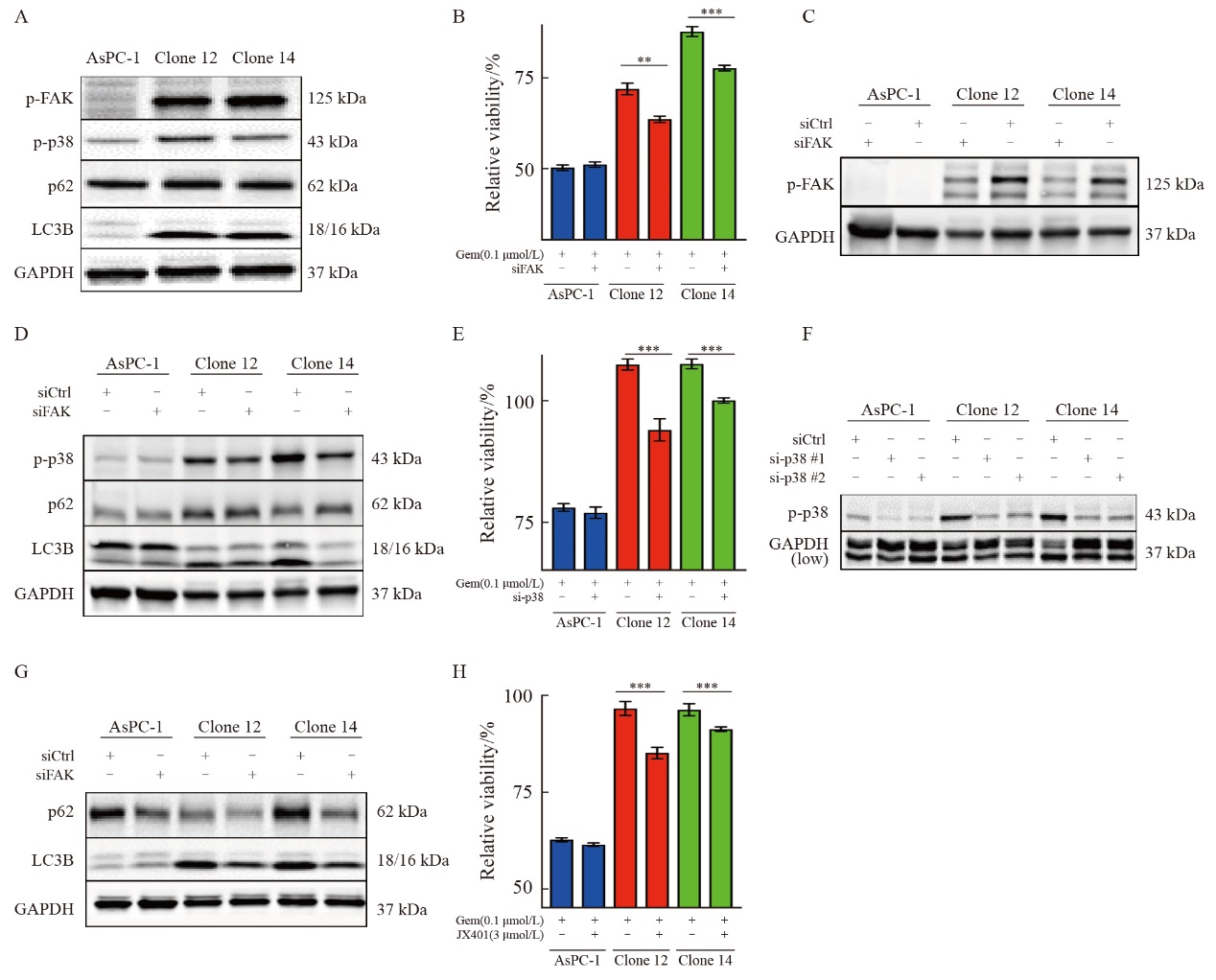
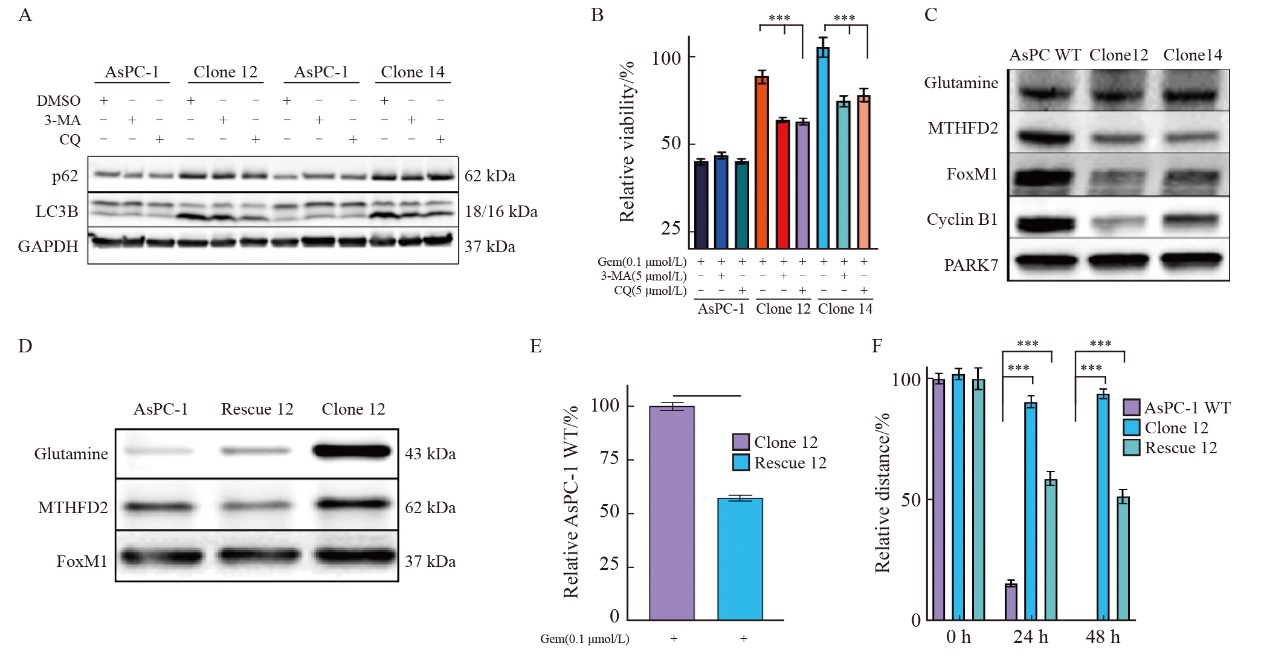
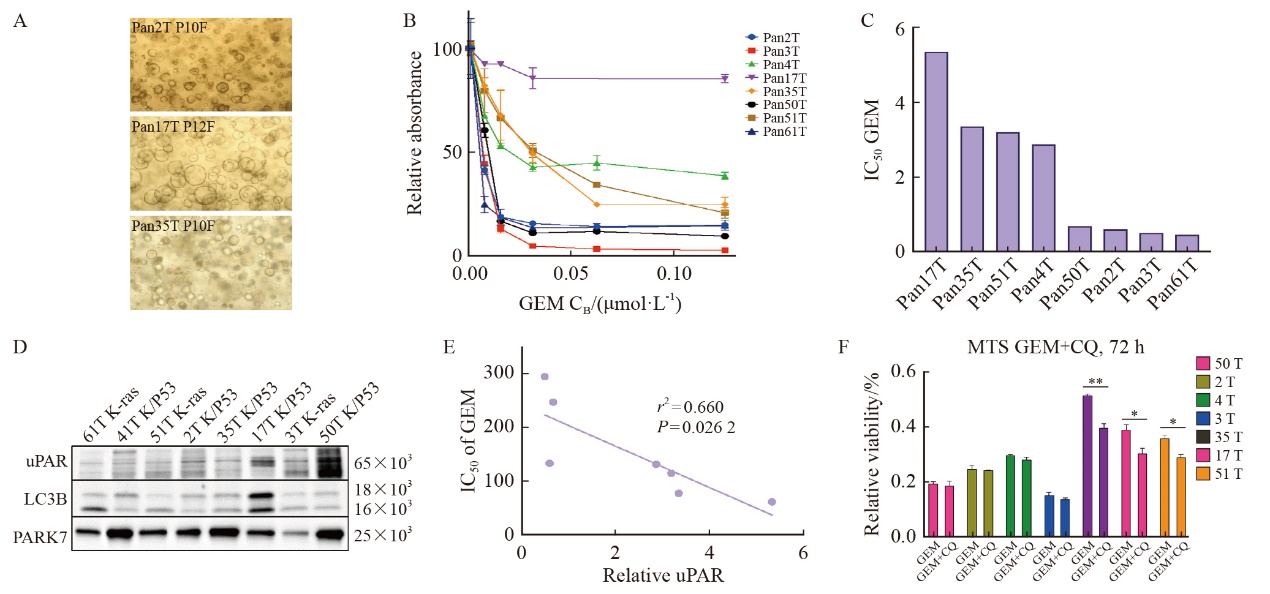
| [1] |
CONROY T, PFEIFFER P, VILGRAIN V, et al. Pancreatic cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up[J]. Ann Oncol, 2023, 34(11): 987-1002.
doi: 10.1016/j.annonc.2023.08.009
pmid: 37678671
|
| [2] |
LIN W, NOEL P, BORAZANCI E H, et al. Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions[J]. Genome Med, 2020, 12(1): 80.
|
| [3] |
PATIL S, STEUBER B, KOPP W, et al. EZH2 regulates pancreatic cancer subtype identity and tumor progression via transcriptional repression of GATA6[J]. Cancer Res, 2020, 80(21): 4620-4632.
doi: 10.1158/0008-5472.CAN-20-0672
pmid: 32907838
|
| [4] |
NOLL E M, EISEN C, STENZINGER A, et al. CYP3A5 mediates basal and acquired therapy resistance in different subtypes of pancreatic ductal adenocarcinoma[J]. Nat Med, 2016, 22(3): 278-287.
doi: 10.1038/nm.4038
pmid: 26855150
|
| [5] |
NEVALA-PLAGEMANN C, HIDALGO M, GARRIDO-LAGUNA I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer[J]. Nat Rev Clin Oncol, 2020, 17(2): 108-123.
|
| [6] |
MIZRAHI J D, SURANA R, VALLE J W, et al. Pancreatic cancer[J]. Lancet, 2020, 395(10242): 2008-2020.
doi: S0140-6736(20)30974-0
pmid: 32593337
|
| [7] |
KUMAR A A, BUCKLEY B J, RANSON M. The urokinase plasminogen activation system in pancreatic cancer: prospective diagnostic and therapeutic targets[J]. Biomolecules, 2022, 12(2): 152.
|
| [8] |
ARONEN A, AITTONIEMI J, HUTTUNEN R, et al. P-suPAR may reflect the inflammatory response after pancreatic surgery[J]. Pancreatology, 2024, 24(1): 146-151.
doi: 10.1016/j.pan.2023.11.006
pmid: 38000982
|
| [9] |
ZHAI B T, TIAN H, SUN J, et al. Urokinase-type plasminogen activator receptor (uPAR) as a therapeutic target in cancer[J]. J Transl Med, 2022, 20(1): 135.
|
| [10] |
PESCE N A, PLASTINO F, REYES-GOYA C, et al. Mitigation of human iris angiogenesis through uPAR/LRP-1 interaction antagonism in an organotypic exvivo model[J]. FASEB J, 2024, 38(5): e23533.
|
| [11] |
SMITH H W, MARSHALL C J. Regulation of cell signalling by uPAR[J]. Nat Rev Mol Cell Biol, 2010, 11(1): 23-36.
|
| [12] |
FERRARIS G M S, SCHULTE C, BUTTIGLIONE V, et al. The interaction between uPAR and vitronectin triggers ligand-independent adhesion signalling by integrins[J]. EMBO J, 2014, 33(21): 2458-2472.
doi: 10.15252/embj.201387611
pmid: 25168639
|
| [13] |
TAN X M, YAN Y H, SONG B, et al. Focal adhesion kinase: from biological functions to therapeutic strategies[J]. Exp Hematol Oncol, 2023, 12(1): 83.
|
| [14] |
ZHENG Y H, XIA Y, HAWKE D, et al. FAK phosphorylation by ERK primes ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST[J]. Mol Cell, 2009, 35(1): 11-25.
doi: 10.1016/j.molcel.2009.06.013
pmid: 19595712
|
| [15] |
ZHENG Y H, LU Z M. Paradoxical roles of FAK in tumor cell migration and metastasis[J]. Cell Cycle, 2009, 8(21): 3474-3479.
pmid: 19829089
|
| [16] |
WU H W, LIANG Z Y, SHI X H, et al. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines[J]. Mol Cancer, 2009, 8: 125.
doi: 10.1186/1476-4598-8-125
pmid: 20021699
|
| [17] |
JI C B, ZHANG M L, HU J J, et al. The kinase activity of integrin-linked kinase regulates cellular senescence in gastric cancer[J]. Cell Death Dis, 2022, 13(7): 577.
|
| [18] |
ZHOU Y H, XIE Y, LUO Y Z, et al. SNAI2 enhances HPV-negative cervical cancer cell dormancy by modulating u-PAR expression and the activity of the ERK/p38 signaling pathway in vitro[J]. Oncol Rep, 2024, 52(2): 104.
|
| [19] |
HILDENBRAND R, ALLGAYER H, MARX A, et al. Modulators of the urokinase-type plasminogen activation system for cancer[J]. Expert Opin Investig Drugs, 2010, 19(5): 641-652.
|
| [20] |
YAO S, PENG L G, ELAKAD O, et al. One carbon metabolism in human lung cancer[J]. Transl Lung Cancer Res, 2021, 10(6): 2523-2538.
|
| [21] |
BOHNENBERGER H, KADERALI L, STRÖBEL P, et al. Comparative proteomics reveals a diagnostic signature for pulmonary head-and-neck cancermetastasis[J]. EMBO Mol Med, 2018, 10(9): e8428.
|
| [22] |
ELAKAD O, HÄUPL B, LABITZKY V, et al. Activation of CD44/PAK1/AKT signaling promotes resistance to FGFR1 inhibition in squamous-cell lung cancer[J]. NPJ Precis Oncol, 2022, 6(1): 52.
|
| [23] |
PENG L G, LI Y C, YAO S, et al. Urokinase-type plasminogen activator receptor (uPAR) cooperates with mutated KRAS in regulating cellular plasticity and gemcitabine response in pancreatic adenocarcinomas[J]. Cancers, 2023, 15(5): 1587.
|
| [24] |
WIŚNIEWSKI J R, MANN M. A proteomics approach to the protein normalization problem: Selection of unvarying proteins for MS-based proteomics and Western blot[J]. J Proteome Res, 2016, 15(7): 2321-2326.
|
| [25] |
SHMAKOVA A A, KLIMOVICH P S, RYSENKOVA K D, et al. Urokinase receptor uPAR downregulation in neuroblastoma leads to dormancy, chemoresistance and metastasis[J]. Cancers, 2022, 14(4): 994.
|
| [26] |
SUGIOKA K, NISHIDA T, KODAMA-TAKAHASHI A, et al. Urokinase-type plasminogen activator negatively regulates α-smooth muscle actin expression via Endo180 and the uPA receptor in corneal fibroblasts[J]. Am J Physiol Cell Physiol, 2022, 323(1): C104-C115.
|
| [27] |
LOGAN R, JEFFERS A, QIN W Y, et al. TGF-β regulation of the uPA/uPAR axis modulates mesothelial-mesenchymal transition (MesoMT)[J]. Sci Rep, 2021, 11: 21210.
doi: 10.1038/s41598-021-99520-5
pmid: 34707211
|
| [28] |
AGUIRRE-GHISO J A, LIU D, MIGNATTI A, et al. Urokinase receptor and fibronectin regulate the ERK (MAPK) to p38 (MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo[J]. Mol Biol Cell, 2001, 12(4): 863-879.
|
| [29] |
LEVY J M M, TOWERS C G, THORBURN A. Targeting autophagy in cancer[J]. Nat Rev Cancer, 2017, 17(9): 528-542.
doi: 10.1038/nrc.2017.53
pmid: 28751651
|
| [30] |
BRYANT K L, STALNECKER C A, ZEITOUNI D, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer[J]. Nat Med, 2019, 25(4): 628-640.
doi: 10.1038/s41591-019-0368-8
pmid: 30833752
|
| [31] |
LA BELLE FLYNN A, CALHOUN B C, SHARMA A, et al. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression[J]. Nat Commun, 2019, 10(1): 3668.
|
| [32] |
BOJ S F, HWANG C I, BAKER L A, et al. Organoid models of human and mouse ductal pancreatic cancer[J]. Cell, 2015, 160(1/2): 324-338.
|
| [33] |
TOKUMO K, MASUDA T, NAKASHIMA T, et al. Association between plasminogen activator inhibitor-1 and osimertinib tolerance in EGFR-mutated lung cancer via epithelial-mesenchymal transition[J]. Cancers, 2023, 15(4): 1092.
|
| [34] |
ZHANG T, WANG B F, SU F, et al. TCF7L2 promotes anoikis resistance and metastasis of gastric cancer by transcriptionally activating PLAUR[J]. Int J Biol Sci, 2022, 18(11): 4560-4577.
doi: 10.7150/ijbs.69933
pmid: 35864968
|
| [35] |
HILDENBRAND R, NIEDERGETHMANN M, MARX A, et al. Amplification of the urokinase-type plasminogen activator receptor (uPAR) gene in ductal pancreatic carcinomas identifies a clinically high-risk group[J]. Am J Pathol, 2009, 174(6): 2246-2253.
doi: 10.2353/ajpath.2009.080785
pmid: 19435784
|
| [36] |
HENKE E, NANDIGAMA R, ERGÜN S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy[J]. Front Mol Biosci, 2019, 6: 160.
doi: 10.3389/fmolb.2019.00160
pmid: 32118030
|
| [37] |
HUANG J Y, LIN Y C, CHEN H M, et al. Adenine combined with cisplatin promotes anticancer activity against hepatocellular cancer cells through AMPK-mediated p53/p21 and p38 MAPK cascades[J]. Pharmaceuticals, 2022, 15(7): 795.
|
| [38] |
YAMAMOTO K, VENIDA A, YANO J, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I[J]. Nature, 2020, 581(7806): 100-105.
|
| [39] |
GANDHARI M, ARENS N, MAJETY M, et al. Urokinase-type plasminogen activator induces proliferation in breast cancer cells[J]. Int J Oncol, 2006, 28(6): 1463-1470.
pmid: 16685447
|
| [40] |
GONIAS S L, HU J J. Urokinase receptor and resistance to targeted anticancer agents[J]. Front Pharmacol, 2015, 6: 154.
doi: 10.3389/fphar.2015.00154
pmid: 26283964
|
| [41] |
SHIBUE T, WEINBERG R A. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications[J]. Nat Rev Clin Oncol, 2017, 14(10): 611-629.
|
| [42] |
MEHNERT J M, MITCHELL T C, HUANG A C, et al. BAMM (BRAF autophagy and MEK inhibition in melanoma): a phase Ⅰ/Ⅱ trial of dabrafenib, trametinib, and hydroxychloroquine in advanced BRAFV600-mutant melanoma[J]. Clin Cancer Res, 2022, 28(6): 1098-1106.
|
| [43] |
JAIN V, HARPER S L, VERSACE A M, et al. Targeting UGCG overcomes resistance to lysosomal autophagy inhibition[J]. Cancer Discov, 2023, 13(2): 454-473.
|
 ), 姚莎2, 王桂华1, 彭罗根1(
), 姚莎2, 王桂华1, 彭罗根1(